
Genetic Epilepsies and Neurodevelopmental Disorders Research Antwerp.
Background
Disorders of brain functioning are heterogeneous, but result in common effects with detrimental consequences in daily life. Disorders of brain functioning can be either developmental or occur later in life, i.e. after a brain hemorrhage or trauma. The main focus of our group is to unravel the genetic causes of developmental brain disorders and more specifically those causing cerebral palsy (CP). CP is the leading cause of motor impairment in children due to damage in the developing brain. It is often accompanied by a wide range of medical conditions such as intellectual disability, learning difficulties, speech and language deficits, epilepsy, autism spectrum disorder and visual and/or hearing impairment. Perinatal oxygen deprivation was long thought to be the leading cause of CP, but recent studies demonstrate this as a cause in at most 12% of patients and genetic causes of CP are increasingly being discovered. However, insights into underlying mechanisms leading to CP are still limited.
A subgroup of disorders that lead to CP are cerebrovascular disorders, e.g. COL4A1- and COL4A2-related disorders. In these disorders, a weakening of the cerebral blood vessels leads to an increased risk of cerebral hemorrhage; this can occur already during pregnancy or in early childhood, but in other patients, the disorder presents only at a later age, e.g. with intracranial aneurysm (widening of the blood vessels of the brain). The underlying mechanism of cerebrovascular disease associated with COL4A1- and COL4A2-variants in both CP and adult-onset intracranial aneurysms is still not fully understood. In addition, studies focused on the identification of additional genetic factors in both infantile and adult cerebrovascular disorders, including intracranial aneurysms, may contribute to unravel underlying pathomechanisms.
Malformations of cortical development (MCD) constitute of a group of rare congenital brain malformations that can also present as CP and associated features. Disorders in the MCD spectrum include lissencephaly (smooth brain), heterotopia (aberrantly located bands or clusters of neurons), and polymicrogyria (multiple small convolutions of the cerebral cortex) with or without microcephaly (small brain) or megalencephaly (large brain). Despite a significant effort in the identification of genetic causes underlying MCD, 60% of MCD cases currently remain molecularly unexplained. Alternative molecular diagnostic approaches are needed to increase the genetic uptake in MCD patients. Additionally, for this subtype of brain disorders, a better understanding of mechanisms involved will be crucial to make steps forward in improvement of care and treatment.
Goal
Our research focuses on deciphering the genetic background and underlying pathomechanisms of these subgroups of brain disorders. We strongly believe that our research contributes to diagnosis and improvement of patient care and can be the entry point for the development of novel targeted therapies.
Strategy
We use state-of-the-art approaches including exome and genome sequencing and transcriptomics to gain insight into the molecular background and pathomechanisms of these groups of disorders. In vivo modelling of COL4A1- and COL4A2-related disorders is also part of our portfolio.
Disorders under investigation
COL4A1- and COL4A2-related disorders, cerebral aneurysm, cerebral palsy, KIF1A-related NESCAV syndrome, malformations of cortical development, mTORopathies, tubulinopathies
Team members
Marije Meuwissen, Anna Jansen, Dorien Schepers, Katrien Janssens, Claudio D'Incal, Liene Thys, Jessica Rosenblum, Ellen Elinck, Lucine Hartuniyan, Ayu Scott
- Cerebrovascular disease and aneurysms research (PhD student Merlijn Nemegeer)
- Brain malformations research (PhD student Jessica Rosenblum)
- Cerebral palsy research (PhD student Liene Thys)
- Mastering Genes of the Brain: Understanding Neurodevelopmental Complexity of Autism-Intellectual Disability Syndromes. (Postdoctoral researcher Claudio D'Incal)
The role of the COL4A2 in cerebrovascular and aneurysmal disorders: a functional approach.
Intracranial aneurysms (IAs) are sac-like dilatations localized in the cerebral artery walls, due to weakening of the vessel wall. Each year 0.7-1.9% of IAs rupture and lead to subarachnoid hemorrhage (SAH). It has a fatal outcome in 35% of affected individuals, a 25% risk of death in the first 10 years after the SAH and permanent brain damage in two-thirds of survivors. Intracranial aneurysms have been described in genetic conditions, i.e., autosomal dominant polycystic kidney disease (ADPKD) or COL4A1- or COL4A2-related disorders, often affecting vascular smooth muscle cell or vascular basement membrane function, strongly suggests additional genetic risk factors for an IA aneurysm formation and rupture.
This project will functionally investigate the contribution of variants in COL4A2 to cerebrovascular and aortic aneurysmal pathology by 1) whole exome sequencing analysis of genetically unsolved patients with intracranial aneurysms, 2) the investigating the effect of COL4A2 variants on ER stress in patient fibroblasts with COL4A2 variants, and 3) establishing and characterizing a col4a2 zebrafish model based on a pathogenic col4a2 helical variant in order to model and functionally validate other col4a2 variants in a second stage.
Our findings will ultimately improve patient care by a better characterization of the phenotypic spectrum, a better interpretation of variants and the identification of potential therapeutic strategies.
PhD student: Merlijn Nemegeer
Promotors: Marije Meuwissen, Bart Loeys & Dorien Schepers
A transcriptome-driven approach for unraveling genetically determined brain malformations.
Malformations of cortical development (MCD) are a heterogenous group of brain malformations, which represent a significant burden for health care and society. Affected individuals suffer from epilepsy and varying degrees of intellectual and motor disability. Using current molecular techniques, over half of MCD cases remain unsolved.
We aim to establish of a gene-specific disease signature based on RNA-sequencing data that pinpoints the disease gene or pathway on which whole genome sequencing (WGS)-based variant analysis should be focused. The selected genes affect either the PI3K-AKT-mTOR pathway or microtubule dynamics, two major pathways involved in brain development. Furthermore, we aim to increase the diagnostic yield in MCD patients by integrating transcriptomics and WGS data of currently "unsolved" MCD cases, allowing the identification of additional variants of interest. Finally, we use the identified disease signatures in the validation of novel MCD candidate genes with similar pathophysiological mechanisms. The results of this project will guide the implementation of transcriptome analysis as another tool in the genetic diagnostic toolbox for MCD and hereby improve patient management and appropriate counseling of families.
PhD student: Jessica Rosenblum
Promotors: Anna Jansen & Marije Meuwissen
Autophagy dysregulation in cerebral palsy: a common mechanism?
Cerebral palsy (CP) is the most frequent cause of motor impairment in children. For long, lack of oxygen at birth has been considered the main cause of CP. However, large population-based studies have shown it to play a role in only around 12% of CP patients, while genetic causes are increasingly demonstrated. To this day, these genetic causes are heterogeneous and common mechanisms of action remain largely unknown. In our research project, systematic genetic analyses in a large cohort of paediatric CP patients led to the identification of multiple genetic variants in genes linked to autophagy. Autophagy is an important biological process that breaks down dysfunctional cell components, facilitating homeostatic balance and reduced neurotoxicity crucial for cell survival in times of stress (e.g. during birth). To assess the exact role of autophagy in the development of CP, we perform different analyses on patient-derived skin fibroblasts. The results of this research may have direct consequences for patient care, leading to improved diagnostic strategies, genetic counseling and even new treatment options in the future.
PhD student: Liene Thys
Promotors: Marije Meuwissen, Berten Ceulemans, Diane Beysen & Katrien Janssens
Mastering Genes of the Brain: Understanding Neurodevelopmental Complexity of Autism-Intellectual Disability Syndromes.
Neurodevelopmental disorders (NDDs) are defined as a group of heterogeneous conditions with early onset in life, affecting the central nervous system, including the development of the brain. NDDs contribute to serious health problems in our society, affecting more than 3% of children worldwide. Patients present with a variety of symptoms and share multiple clinical features such as cognitive impairments, language and speech abnormalities, motor skills delays, learning disorders, and unusual adaptive behaviors, usually diagnosed during infancy, childhood, or adolescence. The clinical presentation is classified according to the Diagnostic and Statistical Manual of Mental Disorders, 5th edition text revision (DSM-5-TR) in intellectual disability, communication disorders, autism spectrum disorder, attention deficit hyperactivity disorder, specific learning disorder and motor disorders. This doctoral work entails two frequent autistic syndromes comorbid with intellectual disability: (1) Helsmoortel-Van der Aa syndrome and (2) fragile X syndrome.
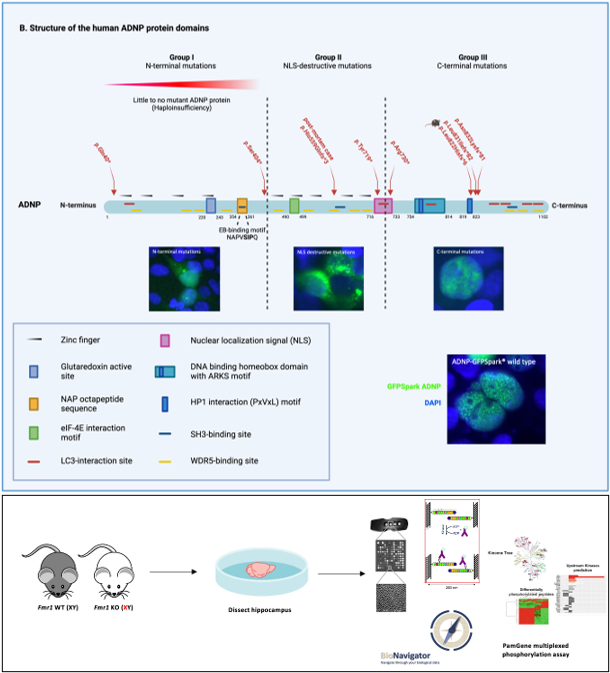
Helsmoortel-Van der Aa syndrome (ADNP)
The development of whole exome sequencing (WES) has substantially increased our insights in the genetic causes of neurodevelopmental disorders by detection of de novo mutations by comparing the exome of the proband to that of its parents. Using this method, mutations in the Activity Dependent-Neuroprotective Homeobox Protein (ADNP) gene have been discovered, contributing to a neurogenetic syndrome termed Helsmoortel-Van der Aa syndrome (OMIM 615873), with a prevalence of 0.2% of global autism cases. Patients show a clinical presentation of mild to severe intellectual disability, autism spectrum disorder, global developmental delay, motor and speech delay, behavior abnormalities and deficiencies in several organ systems such as gastrointestinal problems. In recent screening studies, ADNP appears one of the most frequently mutated genes with a hundred percent disease penetrance. While massive screening studies now have cumulated in the discovery of over a thousand genes that are involved in autism and/or intellectual disability, our molecular and functional understanding of the pathophysiology of these genes is lagging far behind. For instance, despite a wealth of information, many biochemical aspects of the function of ADNP in the brain remain unknown.
Heterozygous ADNP mutations cause incorporation of a premature termination codon in the transcript and escape nonsense-mediated decay, thereby truncating the protein. However, detection of the wild-type protein and ADNP mutants is ambiguous by means of western blotting with multiple laboratories reporting conflicting results. For this reason, we will encompass several detection strategies to characterize the ADNP protein in various specimens. Moreover, truncating ADNP mutants different in expression of the protein and subcellular localization, depending on the location of the ADNP mutation. While these overexpression systems provide a wealth of information, the necessity for an animal model remains imperative to comprehensively study Adnp mutations in the brain. Therefore, we created a novel Adnp heterozyous mouse model containing a C-terminal frameshift mutation akin to a patient mutation. Here, we will perform an extensive molecular and behavioral characterization of our novel mouse model to unravel dysregulated brain pathway and identify targets that hold promise for therapy. Unfortunately, treatments to completely reverse symptoms of Helsmoortel-Van der Aa syndrome patients have currently not been discovered. However, several potential drug candidates have been identified, aiming to alleviate some of the symptoms associated with the condition. Therefore, we will test the experimental ADNP-derived neuroprotective drug candidate NAP/Davunetide in three Adnp heterozygous mouse models with a different mutational origin in hope to reverse part of the dysregulated brain pathways and abnormal behaviors. To further study the Helsmoortel-Van der Aa syndrome, immortalized human cell lines, murine tissues, embryonic stem cells or other model systems have been investigated. However, none of these studies have been performed in disease-relevant tissue. Hence, we will implement a unique case study on autopsy material of a six-year-old child with the heterozygous c.1676dupA/p.His559Glnfs*3 de novo ADNP mutation, thereby substantiating results from published in vitro and in vivo model systems to the human ADNP brain. Taken together, we anticipate that the results of this doctoral project will provide essential insights in the pathogenesis of the Helsmoortel-Van der Aa syndrome, thereby paving the way for therapy for the affected patients.
Fragile X syndrome (FMR1)
The Fragile X syndrome (OMIM 300624) is the leading monogenetic cause of autism and intellectual disability. Patients show distinct clinical features, including behavioral, physical, and cognitive alternations. Fragile X syndrome is caused by a CGG trinucleotide repeat expansion in the Fragile X messenger ribonucleoprotein 1 (FMR1) gene, resulting in a loss of the corresponding FMRP protein due to epigenetic silencing. FMRP is involved in the translational repression of a specific pool of mRNAs that are controlling neurotransmitter signaling and synaptic morphology. Current treatments are developed to intercept at multiple cellular levels amongst which the postsynaptic membrane receptors are the main target. These receptors are key players in signal transduction, and their downstream target proteins are mainly regulated by phosphorylation. Preclinical experiments in animal models were shown to be successful to improve the fragile X phenotype. However, indistinct results are reported in clinical trials, possibly due to a lack of a proper outcome measure. To address this discrepancy, we will investigate the kinome profile to unravel novel fragile X pathways and novel targets for therapy. Therefore, a PamGene® multiplexed phosphorylation assay will be used to measure serine (S), threonine (T) and tyrosine (Y) kinase activity in hippocampal protein extracts of Fmr1 knockout and wild-type littermate mice. In order to confirm these identified kinase-substrate interactions, phosphoproteins will be enriched by TiO2 beads and analyzed by mass spectrometry. We will map these phosphopeptides in canonical pathways, focusing on overlapping pathways identified in the kinase assay. To our knowledge, we will be the first to use a kinomics approach to identify novel targets for therapy of fragile X syndrome.
Teamleden - Cerebrovasculaire aandoeningen en Cerebrale parese

Marije Meuwissen
Staflid Medische GeneticaMarije Meuwissen
Staflid Medische Genetica
Liene Thys
Doctoraatsbursaal, ASOLiene Thys
Doctoraatsbursaal, ASO
Jessica Rosenblum
PhD studentJessica Rosenblum
PhD student
Teamleden - GENERAte
Genetic Epilepsies and Neurodevelopmental Disorder Research Antwerp
Marije Meuwissen
Staflid Medische GeneticaMarije Meuwissen
Staflid Medische Genetica
Katrien Janssens
LabosupervisorKatrien Janssens
Labosupervisor

Claudio D'Incal
DoctoraatsbursaalClaudio D'Incal
Doctoraatsbursaal
Liene Thys
Doctoraatsbursaal, ASOLiene Thys
Doctoraatsbursaal, ASO
Ellen Elinck
LabotechnicusEllen Elinck
Labotechnicus
Jessica Rosenblum
PhD studentJessica Rosenblum
PhD student

Anna Jansen
HoofddocentAnna Jansen
Hoofddocent